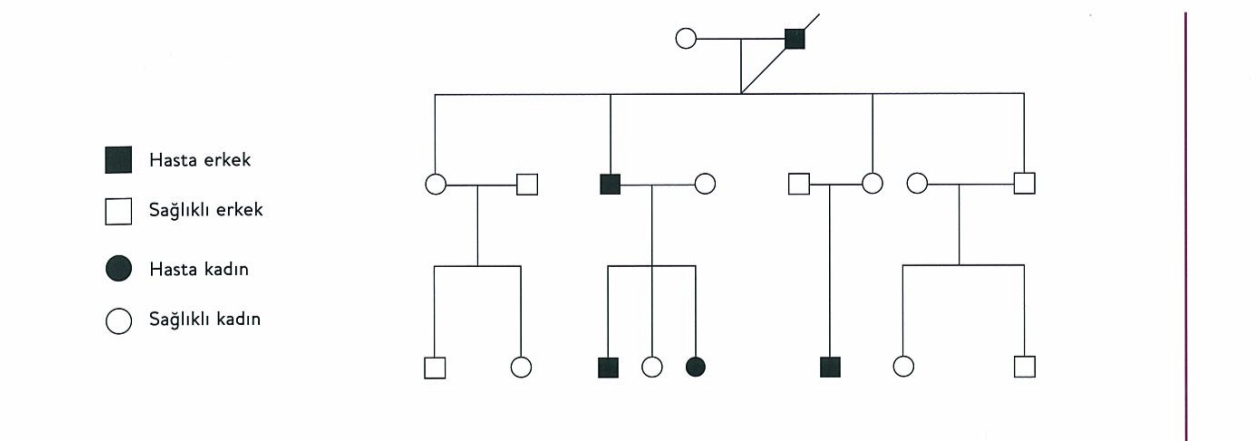
Friedreich ataksisi
Friedreich ataksisi, OR ataksilerin en sık görülen formu olup 25 yaşından önce başlayan ataksilerin % 75ini oluşturur. Friedreich ataksisinde nöropatoloji, dorsal kök ganglionlarındaki nörinopatiye eşlik eden periferik duyusal sinir liflerinde kayıp ve arka kordon dejenerasyonu ile karakterizedir. Hastalık genellikle GAA ekspansiyonuna bağlı olarak ortaya çıkar. GAA tekrar artışı, frataksin proteininin sentezinde bozukluğa yol açmakta bu da mitokondriyal solunum aktivitesini düşürerek hücre içinde serbest radikal birikmesine yol açmaktadır. Azalmış frataksin nörodejeneras
GAA tekrarları ne kadar geniş ise klinik bulgular o kadar ağırdır. Normal alellerdeki tekrar sayısı 6-34¸ dür. Friedreich ataksisinde ise aralık 67-1700 arasında değişmekle birlikte sıklıkla 800-1000 kopyayı içermektedir.
Friedreich ataksisi prevelansı 1/30.000-50.000dir. Hastalık genellikle 20 yaşından önce puberte civarında başlar. Başlangıç yaşı kardeşler arasında bile değişkenlik gösterir.
İlk yakınmalar sık düşme, dengesizlik, yürüme güçlüğü şeklindedir. Ekstremite ve gövde ataksisi gibi serebellar bulgular hastaların hepsinde mevcuttur. Olguların %20'sinde nistagmus görülür. Klinik bulgulara birkaç yıl sonra konuşma bozukluğu (dizartri) eşlik eder. Yutma güçlüğü hastalığın geç dönemlerinde özellikle sıvı gıdalar sırasında ortaya çıkan yakınmalardandır. Vibrasyon ve pozisyon duyusu etkilenmesi olguların yaklaşık % 80inde saptanan bulgulardan biridir
Kortikospinal traktus etkilenmesine bağlı olarak patolojik refleksler ve spastisite ortaya çıkar. Güçsüzlük özellikle alt ekstremitelerde belirgindir. Kas tonusu ise normal veya azalmış olabilir. Hastaların fundoskopik muayenesinde optik atrofi saptanmakla birlikte vizyon kaybı olmayabilir.
İşitme kaybı ve diyabetes mellitus görülebilir. Hastalığın geç döneminde otonomik etkilenmeye bağlı ayak ve bacaklar soğuk ve siyanotik olabilir. Kognitif fonkisyonlar genellikle korunmuştur.
Ayrıca kubbe damak, skolyoz, pes kavus gibi iskelet deformiteleri yanı sıra hipertrofik kardiyomiyopatiye ait klinik bulgular görülebilir. Spesifik bir tedavisi yoktur. Diğer genetik hastalıklarda olduğu gibi daha çok destek tedavi
Spinoserabellar ataksisi
Spinoserebellar ataksiler, çoğunluğu sorumlu genlerdeki mutasyon sonucu gendeki CAG trinükleotid tekrar sayısındaki artışın neden olduğu heterojen bir hastalık grubudur. CAG tekrar sayısındaki artışın saptanması ile bu hastalıkların kesin tanısı konulur.
SCA tip 8'de farklı bir trinükleotid olan CTG tekrar sayısında, SCA 10'da ise ATTCT pentanükleotinin tekrar sayısında artış vardır. Toplumdan topluma değişebilmekle beraber, SCAlar içinde en sık görülenler SCA tip 1,2,3,6,7,10dur. SCA da 1'den 28'e kadar tipler tanımlanmıştır. Toplumda sıklığı yaklaşık 3/100.000 civarındadır.
Spinal-Bulbar musküler atrofisi - kennedy hastalığı androjen reseptör genindeki CAG artışı sonucu oluşur.
Sağlıklı insanlarda 9-37 arsında olan tekrar sayısı hastalarda 40ın üzerindedir.
Progressif güçsüzlük, kas kaybı ile karakterize günlük rutin performansı etkileyen bir hastalıktır. Ortalamatanı yaşı 30-50 yaştır. Tanı konmadan önce ilk yakınmalar kas seğirmesi veya kas krampları olabilir. Ekstremite kaslarına ek olarak çiğneme ve yutma kasları da etkilenebilir.
Androjen duyarlılığında azalmaya sekonder jinekomasti ve testislerde atrofi görülür.
Bu hastalarda kan kreatin kinaz düzeylerinde devam eden bir yükselme vardır. Bu, hastalıkta kas yıkımının önemli bir göstergesidir. Spesifik bir tedavisi yoktur. Kramplar için kinin, baklofen veya E vitamini kullanılabilir.
Arkadaslar aşağıdaki tanımları googleda basit aramayla neolduğunu bulabilirsiniz
Sca1 belirtileri
Primidal bulgular, priferik nöropati,kore
Sca2 belirtileri
Yavaş sakkadlar, periferik nöropati, parkinasyon bulgular-demans akinnetik rijid,kore
Sca3 belirtileri
Primidal ve ekstraprimidal bulgular, nistagmus, amiyotrofi,fasikalasyonlar
Sca4 belirtileri
Duysal aksonal nöropati, sağırlık
Sca5 bertileri
Yavaş seyir, erken başlangıç
Sca6 belirtileri
İzole atksi, yavaş seyir,nistagmus
Sca7 belirtileri
Retinopati, görme kaybı, yüksel oranda antipasyon
Sca 8 belirtileri
Yavaş ilerleyici ataksi, canlı DTR
Sca9 Belirtileri
Bilinmiyor
Sca10 belirtileri
Nöbetler
Sca11 belirtileri
Hafif ataksi
Sca12 Belirtileri
Hipperrrfleksi, ektraprimidal bulgular,demans
Sca13 belirtileri
Mental retardasyon, kısa boy
Sca14 belirtileri
Aksiyal miyokloni
Sca15 belirtileri
Yavaş seyir
Sca16 Belirtileri
Başta tremor
Sca17 belirtileri
Mental yıkım
Sca19 Belirtileri
Kognitif yıkım, miyoklonus,tremor
Sca 20 belirtileri
Dizartri,distoni,dentat kalsifakasyon
Sca21 belirtileri
Hafif kognitif yıkım
Sca22 belirtileri
Yavaş ilerleyici ataksi
Sca23 belirtileri
Geç başlangıçlı ataksi,duysal kayıp
Sca25 belirtileri
Duysal noropati
Sca26 belirtileri
Ataksi,dizatri
Sca27 belirtileri
Diskinezi,kognitif yıkım
Sca28 belirtileri
Nitagmus, ptoz
Frajil x sendromu
Frajil X Sendromu (FXS) kalıtsal zeka geriliğinin bilinen en sık nedenidir. Frajil X sendromlu kişiler zihinsel, davranışsal ve fiziksel bazı farklılıklar gösterirler. Bu sendrom her iki cinsiyeti de etkileyebilir. Kadınlarda yaklaşık 1/250, erkeklerde ise 1/800 kişi FXS na neden olan geni farkında olmadan taşımaktadır. Bu sendrom dünyadaki tüm ırk ve etnik grupları etkiler.
FMR-1 genindeki mutasyon DNA daki CGG tekrarlarının artması şeklindedir. Herkeste FMR-1 geninde yaklaşık 5 ila 40 arasında CGG tekrarı varken, frajil X taşıyıcılarında 55 ila 200 tekrar vardır.
CGG tekrar sayısı frajil X li kişilerde 200den fazladır. Bu genişlemiş tekrar sayısına full (tam) mutasyon denir ve FMR-1 geninin devre dışı kalmasına ve düzgün çalışmamasına neden olur. Devre dışı kalmış bir FMR-1 geni üretmesi gereken proteini yeterli düzeyde ya da hiç üretemez. Proteinin zihinsel gelişim ve işleyiş için kritik önemi vardır.
Frajil X sendromunun klinik özelikleri 3 ana alanda özetlenebilir: Kognitif, fiziksel ve davranışsal. Erkekler kadınlara göre genelde daha ağır etkilenmiştir. Frajil Xe bağlı zeka geriliği erkeklerde yaklaşık 1/3600, kadınlarda 1/4000-6000 görülmektedir.
Kognitif: Zeka düzeyindeki etkilenme normal IQ düzeyi ile birlikte basit öğrenme güçlüğünden ağır kognitif bozukluğa ve otizme kadar geniş bir aralıktadır. Sık rastlanan problemler yürüme, konuşma, tuvalet eğitimi gibi gelişim basamaklarında gerilik, dikkat eksikliği, hiperaktivite ve matematikte zorlanmadır.
Fiziksel: Erişkin erkeklerin genelde uzun yüzleri,büyük ve/veya belirgin kulakları ve büyük testisleri (makroorşidizm) vardır. Düz tabanlık ve kalpte üfürüme neden olacak mitral kapak prolapsusu gibi bağ dokusu ile ilgili problemler sıktır. Kadınlar ve küçük çocuklarda bu özelliklerden bazıları bulunabilir ya da toplumun genelinden farklı olmayabilirler.
Davranışsal: Sosyal ilişki kurabilen arkadaş canlısı bireylerin yanısıra otistik-benzeri davranış gösteren, bazen agresif olanlar da vardır. Frajil X sendromlu kişiler duyusal uyaranlara çok hassastırlar, kalabalığa gürültüye ya da dokunmaya tepki gösterebilirler.
Miyotonik müsküler disrofi
Otozomal dominant kalıtım gösterir ve 1/30.000 sıklığında görülür.
Hem çizgili kasları hem de düz kasları etkiler. Kalp kasları tutulduğundan kardiyak fonksiyonları da bozar. Endokrinopatiler, immün yetmezlikler, katarakt, dismorfik yüz, zekada bozulma ve diğer nörolojik anormallikler eşlik eder.
DNA analizinde 50-2000 arasında değişen CTG tekrarı görülür.
Çoğunlukla yenidoğan döneminde normaldirler.
İlk birkaç yılda güçsüzlük hafiftir. Distal kaslarda progresif erime olur. Dil ince ve atrofiktir. Zaman içinde merdiven çıkma güçleşir ve gowers bulgusu belirginleşir. DTRler korunmuştur. Yürüme becerileri genellikle erişkin yaşta bile kaybolmaz.
Çok nadiren yenidoğan döneminde ağır vakalar görülebilir.
Serum CK düzeyleri normal veya hafif artmış olabilir.
Takipte erken çocuklukta yıllık EKG çekilmeli, eşlik edebilecek endokrin bozukluklar için endokrinolojik değerlendirme yapılmalıdır.
Primer tanı DNA analizinde CTG tekrarlarının gösterilmesidir. Büyük çocuklarda tanı amaçlı kas biyopsisi yararlı olabilir.
Spesifik bir tedavisi yoktur. Ancak kardiyak, endokrin, gastrointestinal ve oküler komplikasyonlar tedavi edilebilir.
Huntington Hastalığı
Dominant kalıtım gösteren MSSnin dejeneratif hastalığıdır. CAG tekrar sayısının arttığı huntington proteinindeki mutasyonlardan kaynaklanır.
Nöron nukleusu içinde anormal inklüzyonlar oluşturarak diğer genlerin transkripsiyonlarının aktive olması için gerekli olan CREB bağlama proteini gibi proteinlerin seviyesini azaltır.
Progresif korea ve presenil demans gibi semptomlar tipik olarak 35-55 yaşları arasında başlar. Vakaların % 1inden azı 10 yaş altı çocuk vakalardır.
Çocukluk yaş grubunda rijidite, distoni ve nöbetle başlangıç görülür.
Çocuklarda mental yıkım ve davranış bozukluğu ön plandadır. Tipik olarak antikonvülzanlara dirençli jeneralize tonik-klonik nöbetler sıktır.
Vakaların % 50sinde serebellar bulgular görülürken % 2sinde okulomotor apraksi görülür.
Çocuklarda daha ağır seyreder. Ortalama yaşam süresi çocuklarda 8, erişkinlerde 14 yıldır.
Nükleus kaudatus ve putamendeki atrofi nedeniyle bilgisayarlı tomografide bifrontal-bikaudal oran azalmıştır.
MRGde putamende intensite artışı görülebilir. Özel bir tedavisi yoktur. Ancak tanı konulur konmaz aileye genetik danışmanlık verilerek ailede yeni vakaların oluşma riski anlatılmalıdır.
Friedreich ataksisi, OR ataksilerin en sık görülen formu olup 25 yaşından önce başlayan ataksilerin % 75ini oluşturur. Friedreich ataksisinde nöropatoloji, dorsal kök ganglionlarındaki nörinopatiye eşlik eden periferik duyusal sinir liflerinde kayıp ve arka kordon dejenerasyonu ile karakterizedir. Hastalık genellikle GAA ekspansiyonuna bağlı olarak ortaya çıkar. GAA tekrar artışı, frataksin proteininin sentezinde bozukluğa yol açmakta bu da mitokondriyal solunum aktivitesini düşürerek hücre içinde serbest radikal birikmesine yol açmaktadır. Azalmış frataksin nörodejeneras
GAA tekrarları ne kadar geniş ise klinik bulgular o kadar ağırdır. Normal alellerdeki tekrar sayısı 6-34¸ dür. Friedreich ataksisinde ise aralık 67-1700 arasında değişmekle birlikte sıklıkla 800-1000 kopyayı içermektedir.
Friedreich ataksisi prevelansı 1/30.000-50.000dir. Hastalık genellikle 20 yaşından önce puberte civarında başlar. Başlangıç yaşı kardeşler arasında bile değişkenlik gösterir.
İlk yakınmalar sık düşme, dengesizlik, yürüme güçlüğü şeklindedir. Ekstremite ve gövde ataksisi gibi serebellar bulgular hastaların hepsinde mevcuttur. Olguların %20'sinde nistagmus görülür. Klinik bulgulara birkaç yıl sonra konuşma bozukluğu (dizartri) eşlik eder. Yutma güçlüğü hastalığın geç dönemlerinde özellikle sıvı gıdalar sırasında ortaya çıkan yakınmalardandır. Vibrasyon ve pozisyon duyusu etkilenmesi olguların yaklaşık % 80inde saptanan bulgulardan biridir
Kortikospinal traktus etkilenmesine bağlı olarak patolojik refleksler ve spastisite ortaya çıkar. Güçsüzlük özellikle alt ekstremitelerde belirgindir. Kas tonusu ise normal veya azalmış olabilir. Hastaların fundoskopik muayenesinde optik atrofi saptanmakla birlikte vizyon kaybı olmayabilir.
İşitme kaybı ve diyabetes mellitus görülebilir. Hastalığın geç döneminde otonomik etkilenmeye bağlı ayak ve bacaklar soğuk ve siyanotik olabilir. Kognitif fonkisyonlar genellikle korunmuştur.
Ayrıca kubbe damak, skolyoz, pes kavus gibi iskelet deformiteleri yanı sıra hipertrofik kardiyomiyopatiye ait klinik bulgular görülebilir. Spesifik bir tedavisi yoktur. Diğer genetik hastalıklarda olduğu gibi daha çok destek tedavi
Spinoserabellar ataksisi
Spinoserebellar ataksiler, çoğunluğu sorumlu genlerdeki mutasyon sonucu gendeki CAG trinükleotid tekrar sayısındaki artışın neden olduğu heterojen bir hastalık grubudur. CAG tekrar sayısındaki artışın saptanması ile bu hastalıkların kesin tanısı konulur.
SCA tip 8'de farklı bir trinükleotid olan CTG tekrar sayısında, SCA 10'da ise ATTCT pentanükleotinin tekrar sayısında artış vardır. Toplumdan topluma değişebilmekle beraber, SCAlar içinde en sık görülenler SCA tip 1,2,3,6,7,10dur. SCA da 1'den 28'e kadar tipler tanımlanmıştır. Toplumda sıklığı yaklaşık 3/100.000 civarındadır.
Spinal-Bulbar musküler atrofisi - kennedy hastalığı androjen reseptör genindeki CAG artışı sonucu oluşur.
Sağlıklı insanlarda 9-37 arsında olan tekrar sayısı hastalarda 40ın üzerindedir.
Progressif güçsüzlük, kas kaybı ile karakterize günlük rutin performansı etkileyen bir hastalıktır. Ortalamatanı yaşı 30-50 yaştır. Tanı konmadan önce ilk yakınmalar kas seğirmesi veya kas krampları olabilir. Ekstremite kaslarına ek olarak çiğneme ve yutma kasları da etkilenebilir.
Androjen duyarlılığında azalmaya sekonder jinekomasti ve testislerde atrofi görülür.
Bu hastalarda kan kreatin kinaz düzeylerinde devam eden bir yükselme vardır. Bu, hastalıkta kas yıkımının önemli bir göstergesidir. Spesifik bir tedavisi yoktur. Kramplar için kinin, baklofen veya E vitamini kullanılabilir.
Arkadaslar aşağıdaki tanımları googleda basit aramayla neolduğunu bulabilirsiniz
Sca1 belirtileri
Primidal bulgular, priferik nöropati,kore
Sca2 belirtileri
Yavaş sakkadlar, periferik nöropati, parkinasyon bulgular-demans akinnetik rijid,kore
Sca3 belirtileri
Primidal ve ekstraprimidal bulgular, nistagmus, amiyotrofi,fasikalasyonlar
Sca4 belirtileri
Duysal aksonal nöropati, sağırlık
Sca5 bertileri
Yavaş seyir, erken başlangıç
Sca6 belirtileri
İzole atksi, yavaş seyir,nistagmus
Sca7 belirtileri
Retinopati, görme kaybı, yüksel oranda antipasyon
Sca 8 belirtileri
Yavaş ilerleyici ataksi, canlı DTR
Sca9 Belirtileri
Bilinmiyor
Sca10 belirtileri
Nöbetler
Sca11 belirtileri
Hafif ataksi
Sca12 Belirtileri
Hipperrrfleksi, ektraprimidal bulgular,demans
Sca13 belirtileri
Mental retardasyon, kısa boy
Sca14 belirtileri
Aksiyal miyokloni
Sca15 belirtileri
Yavaş seyir
Sca16 Belirtileri
Başta tremor
Sca17 belirtileri
Mental yıkım
Sca19 Belirtileri
Kognitif yıkım, miyoklonus,tremor
Sca 20 belirtileri
Dizartri,distoni,dentat kalsifakasyon
Sca21 belirtileri
Hafif kognitif yıkım
Sca22 belirtileri
Yavaş ilerleyici ataksi
Sca23 belirtileri
Geç başlangıçlı ataksi,duysal kayıp
Sca25 belirtileri
Duysal noropati
Sca26 belirtileri
Ataksi,dizatri
Sca27 belirtileri
Diskinezi,kognitif yıkım
Sca28 belirtileri
Nitagmus, ptoz
Frajil x sendromu
Frajil X Sendromu (FXS) kalıtsal zeka geriliğinin bilinen en sık nedenidir. Frajil X sendromlu kişiler zihinsel, davranışsal ve fiziksel bazı farklılıklar gösterirler. Bu sendrom her iki cinsiyeti de etkileyebilir. Kadınlarda yaklaşık 1/250, erkeklerde ise 1/800 kişi FXS na neden olan geni farkında olmadan taşımaktadır. Bu sendrom dünyadaki tüm ırk ve etnik grupları etkiler.
FMR-1 genindeki mutasyon DNA daki CGG tekrarlarının artması şeklindedir. Herkeste FMR-1 geninde yaklaşık 5 ila 40 arasında CGG tekrarı varken, frajil X taşıyıcılarında 55 ila 200 tekrar vardır.
CGG tekrar sayısı frajil X li kişilerde 200den fazladır. Bu genişlemiş tekrar sayısına full (tam) mutasyon denir ve FMR-1 geninin devre dışı kalmasına ve düzgün çalışmamasına neden olur. Devre dışı kalmış bir FMR-1 geni üretmesi gereken proteini yeterli düzeyde ya da hiç üretemez. Proteinin zihinsel gelişim ve işleyiş için kritik önemi vardır.
Frajil X sendromunun klinik özelikleri 3 ana alanda özetlenebilir: Kognitif, fiziksel ve davranışsal. Erkekler kadınlara göre genelde daha ağır etkilenmiştir. Frajil Xe bağlı zeka geriliği erkeklerde yaklaşık 1/3600, kadınlarda 1/4000-6000 görülmektedir.
Kognitif: Zeka düzeyindeki etkilenme normal IQ düzeyi ile birlikte basit öğrenme güçlüğünden ağır kognitif bozukluğa ve otizme kadar geniş bir aralıktadır. Sık rastlanan problemler yürüme, konuşma, tuvalet eğitimi gibi gelişim basamaklarında gerilik, dikkat eksikliği, hiperaktivite ve matematikte zorlanmadır.
Fiziksel: Erişkin erkeklerin genelde uzun yüzleri,büyük ve/veya belirgin kulakları ve büyük testisleri (makroorşidizm) vardır. Düz tabanlık ve kalpte üfürüme neden olacak mitral kapak prolapsusu gibi bağ dokusu ile ilgili problemler sıktır. Kadınlar ve küçük çocuklarda bu özelliklerden bazıları bulunabilir ya da toplumun genelinden farklı olmayabilirler.
Davranışsal: Sosyal ilişki kurabilen arkadaş canlısı bireylerin yanısıra otistik-benzeri davranış gösteren, bazen agresif olanlar da vardır. Frajil X sendromlu kişiler duyusal uyaranlara çok hassastırlar, kalabalığa gürültüye ya da dokunmaya tepki gösterebilirler.
Miyotonik müsküler disrofi
Otozomal dominant kalıtım gösterir ve 1/30.000 sıklığında görülür.
Hem çizgili kasları hem de düz kasları etkiler. Kalp kasları tutulduğundan kardiyak fonksiyonları da bozar. Endokrinopatiler, immün yetmezlikler, katarakt, dismorfik yüz, zekada bozulma ve diğer nörolojik anormallikler eşlik eder.
DNA analizinde 50-2000 arasında değişen CTG tekrarı görülür.
Çoğunlukla yenidoğan döneminde normaldirler.
İlk birkaç yılda güçsüzlük hafiftir. Distal kaslarda progresif erime olur. Dil ince ve atrofiktir. Zaman içinde merdiven çıkma güçleşir ve gowers bulgusu belirginleşir. DTRler korunmuştur. Yürüme becerileri genellikle erişkin yaşta bile kaybolmaz.
Çok nadiren yenidoğan döneminde ağır vakalar görülebilir.
Serum CK düzeyleri normal veya hafif artmış olabilir.
Takipte erken çocuklukta yıllık EKG çekilmeli, eşlik edebilecek endokrin bozukluklar için endokrinolojik değerlendirme yapılmalıdır.
Primer tanı DNA analizinde CTG tekrarlarının gösterilmesidir. Büyük çocuklarda tanı amaçlı kas biyopsisi yararlı olabilir.
Spesifik bir tedavisi yoktur. Ancak kardiyak, endokrin, gastrointestinal ve oküler komplikasyonlar tedavi edilebilir.
Huntington Hastalığı
Dominant kalıtım gösteren MSSnin dejeneratif hastalığıdır. CAG tekrar sayısının arttığı huntington proteinindeki mutasyonlardan kaynaklanır.
Nöron nukleusu içinde anormal inklüzyonlar oluşturarak diğer genlerin transkripsiyonlarının aktive olması için gerekli olan CREB bağlama proteini gibi proteinlerin seviyesini azaltır.
Progresif korea ve presenil demans gibi semptomlar tipik olarak 35-55 yaşları arasında başlar. Vakaların % 1inden azı 10 yaş altı çocuk vakalardır.
Çocukluk yaş grubunda rijidite, distoni ve nöbetle başlangıç görülür.
Çocuklarda mental yıkım ve davranış bozukluğu ön plandadır. Tipik olarak antikonvülzanlara dirençli jeneralize tonik-klonik nöbetler sıktır.
Vakaların % 50sinde serebellar bulgular görülürken % 2sinde okulomotor apraksi görülür.
Çocuklarda daha ağır seyreder. Ortalama yaşam süresi çocuklarda 8, erişkinlerde 14 yıldır.
Nükleus kaudatus ve putamendeki atrofi nedeniyle bilgisayarlı tomografide bifrontal-bikaudal oran azalmıştır.
MRGde putamende intensite artışı görülebilir. Özel bir tedavisi yoktur. Ancak tanı konulur konmaz aileye genetik danışmanlık verilerek ailede yeni vakaların oluşma riski anlatılmalıdır.
Son düzenleme: